
0 Reflections on the Devonport Incident and the Clothier Report of 1972
Over 50 years ago on the 12th July 1972, Dr Shirley Summerskill (MP for Halifax) spoke to the house of commons and said: - " It is to be deplored that human failings were the main cause of the tragedy, [in March 1972] but it is hoped that all who are concerned with the preparation of infusion fluids will take a lesson from this case. I include in that remark the management and the laboratory staff." She was of course referring to the report of the Committee which Sir Keith Joseph, Secretary of State had instructed, under the chairmanship of Mr. C. M. Clothier, QC, to inquire into the circumstances which led to the use of contaminated dextrose infusion fluids at Devonport Hospital and which involved the death of five patients. The Committee concludes—in its own words in paragraph 79 - " … that the fundamental cause of this disaster is to be found in human failings at Evans Medical, ranging from simple carelessness to poor management of men and plant. The Committee heard of no imminent technological advance in the field of production of intravenous fluids which will eliminate the need for skillful men devoted to their work. The Committee considers that too many people believe that sterilization of fluids is easily achieved with simple plant operated by men of little skill under a minimum of supervision, a view of the task which is wrong in every respect" In 2019 David Churchward a GMP inspector for the MHRA, reflecting on the Devonport incident concluded the following - "Despite significant progress in automation, there is still no replacement for a skilled and dedicated workforce who understand the importance of their work. Industry and regulators must maintain the highest levels of quality culture vigilance." "MHRA inspectors look for indicators showing that personnel at all levels of the organisation have appropriate technical knowledge to enable good decision-making and understand how their actions impact the product and patient." In his article on Quality Culture he went on to say:- A strong quality culture is built upon: knowledge of what is important, and how a process achieves critical quality attributes diligence, by fostering awareness that everyone contributes to product quality, and understanding that “my actions impact the patient and the company” vigilance by individuals who know what ‘right’ and ‘wrong’ look like in their process, and a mechanism for management to be aware of problems senior management commitment to being visible and transparent in decision-making so that positive outcomes can be seen from the diligence and vigilance efforts. This is more than the company mission statement – it’s ‘walking the talk’”. In the aftermath of the Clothier report being published the Medicines Inspectorate increased the number of inspectors, but they accepted that inspection alone would not be enough to prevent a recurrence. This position remains the same today as it did then. Around the time of the incident following a history of non-sterile products in the 1960’s the role of the regional steriliser engineer was created. Charged with the responsibility for providing technical expertise and governance to the sterilisers in their charge. This role continues today in the capacity of the Authorising Engineer (Decontamination), [AE(D)] whose role and responsibility is defined in HTM01-01 part A. [1]. They form the Decontamination Technical Platform, which constitutes registered AE(D)'s who have undergone a rigorous training framework at Eastwood Park and undergo regular peer review to ensure the high standards and expectations of IHEEM and the DTP are maintained. The DTP contributes significantly to the standards and guidance of healthcare and industry through the BSI CH198 committee for sterilization standards development; brining decades or knowledge and experience at a national and international level. [2]. In 1970 NHS Falfield (now Eastwood Park) started residential training courses for hospital engineers. This included training Test Person (Sterilisers) and the regional steriliser engineers were instrumental in the training and sharing of best practise and knowledge. This role exists today as a Competent Person (Decontamination) being defined in HTM01-01 part B, as well as the role of the Authorised Person (Decontamination), the latter responsible for the management of the decontamination and sterilisation service.[3]. Both roles are City & Guilds Accredited at Eastwood Park where AE(D)’s continue to provide robust training for hospital engineers and engineers from industry. These roles within the Healthcare setting provide assurance that the people charged with the testing of sterilisers and their required oversight understand the fundamentals of steam sterilisation and the validation, monitoring and routine control that must be in place to provide sterility assurance. In reflection of the findings of the Clothier report 50 years ago, how confident are you in the training, competency and knowledge of the people involved in the qualification of your sterilisers and in their ongoing routine monitoring and control? Whilst the above roles are clearly defined in the UK Healthcare sector, what about Medical Devices and Pharmaceutical and how robust is your Quality Culture? [1] IHEEM Decontamination Technical Platform - IHEEM [2] Health Technical Memorandum 01-01. Part A: Management and provision (england.nhs.uk) [3] Health Technical Memorandum 01-01. Part B: Common elements (england.nhs.uk)

0 Warning Letter - Failure to adequately establish a procedure governing inspection and acceptance of Biological Indicators
An interesting FDA Warning Letter concerning the inspection and acceptance procedures for Biological Indicators, used in the site sterilization process. 11th October 2024 New Zealand Failure to establish and maintain procedures for acceptance activities, which include inspections, tests, or other verification activities, as required by 21 CFR 820.80(a). For example: 1) Your firm's current incoming inspection and acceptance procedure for Biological Indicator (b)(4) (BI) is not adequately established. This inadequacy could compromise the sterilization process, potentially affecting the safety and effectiveness of the NeoZoline Ventilation Tubes. Your firm discards the Certificate of Conformance provided by (b)(4) with each BI batch, which was requested by the FDA investigator during the inspection. It appears that your firm failed to retain critical documents such as the reconciliation sheet, production batch record, or (b)(4) Certification of Conformance, which are required under your own WI22 "Production QC and Release Product Register," specifically section 2.3, to demonstrate that the product was successfully sterilized. The inspection revealed that your firm does not perform BI Population verification tests on the received BI batches to ensure that the population has not changed during shipping or storage. This is a critical step is necessary to provide a high degree of assurance that the BIs are viable to support verification that finished product is successfully sterilized through the (b)(4) Sterilization process. Additionally, the inspection revealed that critical information such as the batch number and expiration date of the BI used in the sterile load is not documented on the post-sterilization B1 Test Reports, which are used as part of the finished device acceptance and release process. We reviewed your firm's response dated June 13, 2024, and conclude that it is not adequate. Your firm's response indicates that its Initial Action was completed on June 1, 2024. Your firm concluded that a risk analysis of the specific observation indicates no risk to end users for products currently in distribution and manufacturing. That conclusion is based on the following factors: The (b)(4) Biological Indicator is an off the shelf product from supplier where (b)(4) has claimed compliance with ISO 11138-1:2017 and ISO 11138-2:2017. The BI is packaged in a foil pouch designed to protect the product from humidity excursions. The pottles are used to store non-sterile Ventilation Tubes hence the sterility of the pottle is not a requirement. Additionally, your firm has outlined a plan with the following actions to be completed by December 1, 2024, which includes the following actions: (b)(4) Based on your firm's response there is an ambiguity in the definition of the term `pottle' and whether it is required to be sterile or not. (b)(4) your firm's response states that there is no risk for the sterile products manufactured at your facility because the BIs that they use conform to ISO 11138-1:2017 and ISO 11138-2:2017, and that they are in their sterile packaging until they are needed. However, the inspection demonstrated that your firm does not do any testing to verify that the BIs purchased for the purpose of demonstrating that terminal sterilization is achieved, are actually viable when they are used. Additionally, the inspection revealed that they do not capture key data such as BI batch number and expiration date, and thus if a BI falsely indicated that a sterilization was successful, there is no mechanism by which your firm can trace the lots of sterile products back to a failed BI. Additionally, your firm did not include documentation or evidence of the corrections or corrective actions because they are in progress and have not been completed. Additionally, given the observations described above, your firm has not provided a commitment to conduct a retrospective assessment of all finished devices whose sterilization was verified using Biological Indicator (b)(4) to determine whether sterility was compromised.

0 Warning Letter - Failure to sterilize direct product contact items.
Another FDA Warning letter for a facility in Jordan. This is an interesting observation indeed. Published in February of this year, it highlights a deficiency in the understanding of the fundamentals of aseptic sterile manufacturing. 14th Feb 2024 Jordan Your firm failed to establish and follow appropriate written procedures that are designed to prevent microbiological contamination of drug products purporting to be sterile, and that include validation of all aseptic and sterilization processes (21 CFR 211.113(b)). Sterilization of All Equipment that Contacts Sterile Product Constituents Your firm failed to sterilize multiple pieces of equipment (e.g., hoppers, tracks) that directly contacted (b)(4) constituents of your sterile products, including primary containers and closures. It is essential that sterile equipment is used in the processing of all elements of the sterile product. Conducting only cleaning and disinfection (e.g., (b)(4)) of such equipment is inappropriate, as it is not equivalent to use of (b)(4) (or equivalent sterilization method, such as (b)(4)) sterilization of equipment. Validation of sterilization cycles is based on sterility assurance evaluations that are comprised of rigorous physical and biological measurements. In addition, it is essential that only sterile instruments are used for aseptic processing line manipulations and in handling of sterilized materials. Between uses, sterile instruments are held under ISO 5 conditions and continuously maintained in a manner that prevents contamination. It is not acceptable to touch the sterile (b)(4) during aseptic production in the manner performed by your personnel. Annex 1 requires us to ensure that all product contact components and equipment should be sterilized prior to use. This includes raw materials and intermediates and/or bulk solutions, (clause 8.11). The sterilization process should be validated, both physically, with temperature sensors and where appropriate biologically with a suitable biological indicator challenge. The whole of the load should be subject to the required sterilant exposure , (clause 8.36). To ensure this and to deliver a reliable and repeatable sterilisation process the whole of the load should equilibrate to temperature before the sterilization exposure phase commences, (clause 8.52). If it is not possible to assemble or connect load items aseptically within the Grade A core (Grade B background) then sterilization in place validation process needs to be considered. Preferably intrinsic sterile connection devices should be designed to mitigate risk of contamination, (clause 8.14). For materials, items, or components necessary for aseptic processing, that are not direct/indirect product contact then validated disinfection and transfer protocols should be in place, they should be protected from recontamination and the pathways for potential contamination should be clearly risk assessed and included within the EM programme, (clause 8.49) Where items are sterilized in sealed packaging, the packaging must be compatible with the load items and the sterilization process. The sealing process should be validated to ensure integrity post sterilisation. Validation should consider integrity of the sterile barrier system and the maximum hold time prior to sterilisation established and the shelf life assigned to each load item or component should be established, (clause 8.48). Maximum holding times for equipment and component cleaning, drying and sterilization should be documented and validated. The maximum holding time for sterilized equipment and components can be stored prior to use and during filling should be established and validated, (clause 8.18) & (clause 8.46) The qualification of the loading configuration and of the wrapping, orientation and location of individual load items should be carefully considered and clearly documented, such that their unwrapping to enable assembly with other direct and/or indirect product contact parts can be achieved aseptically, (clause 8.12). For moist heat porous hard goods loads, items to be sterilized must be clean, dry and packaged into their protective barrier systems, that comply to relevant parts of ISO 11607. Orientation to allow effective air removal and rapid steam penetration should be ensured, (clause 8.61). Equally, condensate drainage and confirmation the load items are ‘sensibly dry’ upon cycle completion should be verified both in qualification of the load and in routine operation, (clause 8.56).

0 Warning Letter - Failure to define Load Configurations
I like to keep an eye on FDA Warning letters, especially for anything related to sterilization process or equipment issues. This one has recently popped up, dated 5th June 2024. 1. Failure to establish and follow appropriate written procedures designed to prevent microbiological contamination of drug products purporting to be sterile, including procedures for validation of all aseptic and sterilization processes [21 CFR 211.113(b)]. For example: a. The aseptic processes used to manufacture your products have not been validated (e.g., by performing media fill simulations). These products purport to be sterile and are expected to be sterile. b. The (b)(4) sterilization process used for equipment that comes into direct contact with your products has not been adequately validated. For example, you have not defined the optimal parameters and conditions, including the load configuration for sterilizing equipment such as the small instruments (e.g., forceps), glass beakers, and sometimes PPE, used in aseptic processing of your products. This for me is really an own goal for this company, (based in the US). GMP expectations in this matter are quite clear: - In PDA Technical Report No.01, (section 4.4.1.3) it requires porous load patterns to be established after OQ prior to beginning PQ, ensuring items are loaded within the useable chamber space and orientated to aid air removal, steam penetration and condensate drainage, etc. Loading instructions should be documented. In the EU, Annex 1, validated loading patterns should be established for all sterilisation processes and load patterns should be subject to periodic revalidation. Maximum and minimum loads should also be considered as part of the overall load validation strategy, (clause 8.38). Routine operating parameters should be established and adhered to for all sterilisation processes, e.g. physical parameters and loading patterns, (clause 8.40) There should be mechanisms in place to detect a sterilisation cycle that does not conform to the validated parameters. Any failed sterilisation or sterilisation that deviated from the validated process (e.g. have longer or shorter phases such as heating cycles) should be investigated, (clause 8.41). Need help in defining and qualifying your sterilization load configurations? Worried you may not comply. Reach out to me here and avoid an audit observation.

0 NSAI - Working Group 3 on ISO 19253
It was great to attend the NSAI plenary event last week and work on ISO 19253, the new standard for moist heat sterilization of contained products. The plenary event is taking place from June 24th to June 28th at the Atlantic Technological University Galway City Campus. ISO/TC 198 is responsible for specifying requirements for #cleaning, #disinfecting, #sterilizing and aseptic processing of #health care products together with associated equipment and ancillary products used in ensuring effective application of these processes. ISO TC 198 is also responsible for harmonizing terminology related to its scope of work. This event will see the gathering of #global #experts in the field in the pursuit of the further development of standards in this area.Read more about the event here - https://lnkd.in/ehwVWmQB
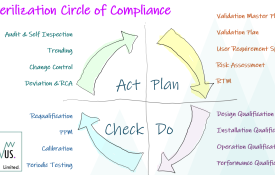
0 Sterilization Process & Equipment Validation: Circle of Compliance
Many are familiar with the Validation V-Model and the Deming Wheel. Like the Reusable Instrument Cycle in HTM01-01 [1] I like to visualize the Validation Lifecyle as a circular journey, moving through each of the 4 Stage Gates of Plan-Do-Check-Act. Within each stage there are discreet activities that we must complete and over the coming weeks I will go through each stage and discuss the essential elements that one must consider in the Commissioning, Validation, Routine Monitoring & Control of your sterilization process or equipment.For me, the validation exercise is not a journey along a one way street, culminating in annual Requalification testing, but a circle of compliance measures, checks and balances put in place to maintain our compliance position and sustain a repeatable and reliable service. [1] HTM01-01: Part A - Management & Provision, Clause 1.5.

0 New publication: BS EN ISO 17665:2024
Moist heat sterilization just got easier. BS EN ISO 17665:2024 is the newly published and radically revised international standard on steam sterilization processes for medical devices. It’s now fully up to date and utterly comprehensive.To comprehend the major changes, read our free of charge Executive Briefing. It breaks down BS EN ISO 17665:2024 into bite-sized, actionable insights that explains what the standard covers, and the changes that have been made, challenges and benefits of using it. Navigating the complex regulatory landscape is a key aspect of sterilization validation. Kevin's in-depth knowledge of global regulations, including FDA, ISO, and industry-specific guidelines can assist you in developing effective moist heat sterilization validation strategies, preparing regulatory submissions, and ensuring compliance with the applicable standards. With his guidance, you can navigate the regulatory processes smoothly and efficiently, saving time and resources whilst meeting the necessary requirements. Download now: https://bit.ly/3ycqOqxOr shop today: https://bit.ly/4bnU0cy#BSIStandards hashtag#ISO17665 #MedicalDeviceSterilization #MoistHeatSterilization #MedicalDevices
0 New Publication: BS EN ISO 11607-1:2020+A1:2023
The importance of BS EN ISO 11607-1:2020+A1:2023.BS EN ISO 11607-1:2020+A1:2023 can help users to develop their expertise in terminal sterilization, while increasing confidence in their medical devices.This standard helps improve efficiency and strengthen their risk management, alongside safeguarding product quality and patient well-being. Navigating the complex regulatory landscape is a key aspect of sterilization validation. Kevin's in-depth knowledge of global regulations, including FDA, ISO, and industry-specific guidelines can assist you in developing effective moist heat sterilization validation strategies, preparing regulatory submissions, and ensuring compliance with the applicable standards. With his guidance, you can navigate the regulatory processes smoothly and efficiently, saving time and resources whilst meeting the necessary requirements.View this standard on BSI Knowledge today, click below for more.#BSIStandards #BSENISO11607 #MedicalDevices