
0 Thermometric Small Load Test
This is a test for steam penetration into a standard test pack. This test pack is used to identify A level of air removal or reduction in the level of NCGs in the steam within the chamber sufficient to qualify the sterilisation process for a wide range of cannula, metal and textile products. For this test a number of temperature sensors (typically 5 sensors) are located at different levels within the standard test pack around the vertical axis. ISO 17665:2024 - Annex C.4.1 Introduction This test is used to demonstrate that after the air removal stage of the operating cycle, sterilizing conditions are obtained within the chamber and standard test pack. The more air there is to remove, the more exacting will be the test; that is why the pack is used by itself in an otherwise empty chamber. Temperatures and pressures should be recorded by independent measuring equipment. Test Procedure Place a standard test pack in the chamber with the bottom of the pack supported 100-200 mm above the centre of the chamber base. Place three temperature sensors in the following positions: one in an active chamber discharge; five within the test pack as detailed in BS EN 285 (the wire from the sensors should be carefully arranged to prevent steam tracking along it); one placed in the free space 50 ± 5 mm above the approximate centre of the upper surface of the test pack. Connect a pressure recorder (or test gauge) to the chamber. Start the operating cycle, with standard drying time, and take readings as described for the automatic control test. If a test pressure recorder is being used, measure the chamber pressure at the approximate mid-point of the holding time. The test should be considered satisfactory if the following requirements are met: the requirements of the Automatic Control Test are met; during the plateau period the temperature measured above the test pack does not exceed the temperature measured in the active chamber discharge by more than 5°C for the first 60 s and 2°C for the remaining period; the equilibration time determined from the measured temperatures does not exceed 15 seconds for chambers up to 800L and 30 seconds for larger chambers; the holding time determined from the measured temperatures is not less than that specified for the selected sterilization temperature; during the holding time the temperatures measured in the reference measurement point and in the centre of the test pack: are within the appropriate sterilization temperature band; do not fluctuate by more than ± 1°C; do not differ from one another by more than 2°C; during the holding time: the indicated and recorded chamber temperatures are within 1°C of the temperature measured in the reference measurement point; the indicated and recorded chamber pressures are within 0.05 bar of the measured pressure; for sterilizers using vacuum as the sole method of drying: the duration of the drying stage is not less than 3 minutes; the chamber pressure at the end of the stage does not exceed 40 mbar absolute; at the end of the cycle the sheets are sensibly dry. Return to Schedule of Tests
0 Bowie & Dick Steam Penetration Test
This test is a steam penetration test, similar to the small load test and intended for daily use. This test is also used to identify a level of air removal or reduction in the level of non-condensable gases in the steam within the chamber sufficient to qualify the sterilisation process for a wide range of cannula, metal and textile products. A chemical indicator meeting the requirements of 11140-3 is placed in the centre of a standard test pack and a pass is identified from a uniform colour change to the indicator. The standard test pack described, offers a challenge to the sterilisation process nominally the same as the challenge from the textile test pack described by Dr Bowie. Indicators conforming with 11140-4 can be used as an alternative to the standard test pack for conducting the Bowie and Dick steam penetration test. ISO 17665:2024- Annex C.5 NOTE - The rate of steam admission to the chamber during pressurisation to the sterilisation stage can have an influence on the measured thermometric values or the visible change taking place in the chemical indicator. Thus, if the rate of pressurisation is slow then any residual air pocket can be heated close to steam temperature leading to an inaccurate determination of temperature difference between the drain and the centre of the test pack. Similarly, any residual air present can be sufficiently heated and humidified during slow rate of pressurisation to allow a visible change in a chemical indicator placed within the test pack. Introduction Sterilization is achieved by the rapid and even penetration of steam into all parts of the load and the maintenance of these conditions for the specified holding time. To ensure this, it is essential to remove air from the chamber and load, and to provide a steam supply which contains a minimal volume of non-condensable gases. Any residual air and non-condensable gases will become concentrated as a bubble in the load and inhibit steam penetration. The Bowie-Dick test shows whether or not steam penetration of the test pack is even and rapid, and thus by implication that air or other non-condensable gases are not present. It does not confirm that the sterilization conditions in the load have been achieved. Principle of the test The test, as originally conceived and described in earlier editions of HTM 10 (Bowie, Kelsey and Thomson, 1963), is based on the use of a chemical indicator in the form of an adhesive tape stuck to a piece of suitable paper to form a St Andrew’s cross. This indicator paper is placed at the centre of a test pack of folded huckaback towels and then subjected to an operating cycle. The indicator tape shows a change of colour in response to a combination of time, temperature and moisture. If no air is present in the chamber, steam will penetrate rapidly and completely, and the indicator will show a uniform colour change. If air is present, it will collect within the pack as a bubble. The indicator in the region of the bubble will be of a different colour than elsewhere on the paper, because of a lower temperature, lower moisture level or both. The modern Bowie-Dick test uses a Class B chemical indicator conforming to BS EN 867: Part 3 contained within a standard test pack. The indicator is distributed over an A4 paper sheet in the form of a geometric pattern. When used in conjunction with a standard test pack, Class B indicators are designed to show a failure either if, at the start of the holding time, the temperature at the centre of the test pack is 2°C or more below the temperature in the active chamber discharge; or if the indicator is exposed to insufficient moisture. Both conditions are usually caused by the presence of air or other non-condensable gases. Because of the tolerances necessary in the manufacture of chemical indicators, users should be aware that in order to detect a temperature difference of 2°C the indicator may show signs of failure with a smaller temperature difference. Test Procedure The Bowie-Dick test is normally preceded by a warm-up cycle. This cycle is necessary because the effectiveness of air removal may depend on all parts of the sterilizer being at working temperature. A satisfactory sterilizer may give a fail result if this is not done. Remove the wrapping from a standard test pack and place the indicator paper in the sheet located nearest to the centre of the pack. Reassemble and secure the pack and replace the wrapping. Place the test pack in the chamber with the bottom of the pack supported 100 to 200 mm above the centre of the chamber base. Select the Bowie-Dick test cycle. Ensure that the holding time will not be longer than that specified in the table below. If this time is exceeded, the indicator may change in such a way as to make it difficult to detect the variations that would indicate a fail condition. Start the operating cycle. During the holding time, note the reading on the cycle counter, the chamber temperature indicator and the chamber pressure indicator. When the cycle is complete, remove the indicator paper from the test pack. The test should be considered satisfactory if the following requirements are met: there is a uniform change throughout the indicator; the automatic controller indicates that a Bowie-Dick test cycle has just been completed. It is important to compare the colour of the indicator at the corners of the paper with that at the centre so that any difference can be clearly seen. If there is any discernible difference the test should be recorded as failed, and the paper marked accordingly. A large area of unchanged indicator points to a gross failure. An unsatisfactory test result indicates that the machine should not be used until the fault has been rectified. It is important to realise that if a sterilizer fails to pass the Bowie-Dick test it cannot be made safe simply by increasing the holding time until a uniform colour change is produced. A failed sterilizer is in urgent need of skilled attention. Several factors may inhibit steam penetration and cause the test to fail. Common causes of failure include the following: a. an inefficient air removal stage; b. an air leak during the air removal stage; c. the presence of non-condensable gases in the steam supply. A subsequent Thermometric Test Small Load will assist in diagnosing the cause of failure: if the test reveals a temperature depression at the centre of the test pack, the problem is likely to be inefficient air removal or an air leak into the chamber. Air remaining in the centre of the test pack is inhibiting the penetration of steam and the correct temperature is not being attained. The sterilizer should not be returned to service until it has been subjected to a vacuum leak test and an air detector function test; if the test fails to reveal a temperature depression, the problem is almost certainly air or other non-condensable gases in the steam supply. In this case the correct temperature is being attained but the steam is diluted, and insufficient moisture is present to change the indicator. The sterilizer should not be returned to service until the steam supply has been tested for the presence of non-condensable gases. Return to Schedule of Tests

When qualifying our autoclave what tests should we do? ISO 17665:2024 (Annex C) includes guidance on recommended periodic tests for moist heat sterilization processes. This post will outline what tests are recommended, when they are suggested to be completed and at what ongoing periodic frequency they should be repeated. Further posts will go into more detail on how these tests are performed and why. The recommended schedule of tests has existed for many years, developing, and changing over time. Published in the 1960’s, HTM 10 first outlined routine tests in Chapter IX, Routine Testing, recommending the Bowie and Dick Test (Daily), Leak Rate Test (at least weekly) and if fitted, sensitivity of the air detector in accordance with BS 3970-1:1966 (as amended in 1969) to require: - “… a device capable of detecting the presence of air in sufficient quantity to prevent the attainment of and maintenance of the sterilizing conditions within the load …. fitted as part of the automatic control.” HTM 10 was subsequently replaced with HTM 2010, that many of us will be familiar with and the schedule of validation and periodic tests within. HTM 01-01 replaced HTM 2010 and is now the current guidance in UK healthcare. Broadly speaking testing falls into two parts. Firstly, the Schedule of Validation Tests outlined what tests should be conducted during Installation, [IQ] Operational [OQ] and Performance Qualification [PQ]. Secondly, the Schedule of Periodic Tests outlines ongoing routine tests and their frequency, nominally conducted daily, weekly, quarterly and annually, (Requalification [RQ]). Following the test regime will ensure that you are in control of your sterilization process and indeed meet the requirements of Annex 1. Read more here: Sterilization Control Strategy Many of these tests and frequencies have been carried forward into ISO 17665:2024 and I would consider them to be Good Autoclave Practice. The tables below outline the recommended tests from the standard: - Schedule of Validation Tests Installation tests Safety tests and checks Operation Qualification (commissioning) Steam Quality Non-Condensable Gases Steam Quality Dryness Value Steam Quality Superheat Steam Quality Contaminants Thermometric Test Small Load Thermometric Test Full Load Hollow Load Test Bowie and Dick Test Air Leakage Flowrate Test Air Detector Small Load Air Detector Full Load Air Detector Function Load Dryness Small Load Textiles Load Dryness Full Load Textiles Load Dryness Metal Load Automatic Control Test Performance Qualification Bowie and Dick Test Automatic Control Test Thermometric Product Load Test Air Leakage Flowrate Test Schedule of Periodic Testing Daily Bowie and Dick Test Weekly Automatic Control Test Bowie and Dick Test Air Leakage Flowrate Test Air Detector Function Three monthly (Quarterly) Automatic Control Test Bowie and Dick Test Air Leakage Flowrate Test Air Detector Function Thermometric Test Small Load Annually Air Leakage Flowrate Test Automatic Control Test Bowie and Dick Test Thermometric Test Small Load Thermometric Test Full Load Air Detector Small Load Air Detector Full Load Air Detector Function Thermometric Product Load Test Steam Quality Non-Condensable Gases Steam Quality Dryness Value Steam Quality Superheat Steam Quality Contaminants

0 Warning Letter - Failure to sterilize direct product contact items.
Another FDA Warning letter for a facility in Jordan. This is an interesting observation indeed. Published in February of this year, it highlights a deficiency in the understanding of the fundamentals of aseptic sterile manufacturing. 14th Feb 2024 Jordan Your firm failed to establish and follow appropriate written procedures that are designed to prevent microbiological contamination of drug products purporting to be sterile, and that include validation of all aseptic and sterilization processes (21 CFR 211.113(b)). Sterilization of All Equipment that Contacts Sterile Product Constituents Your firm failed to sterilize multiple pieces of equipment (e.g., hoppers, tracks) that directly contacted (b)(4) constituents of your sterile products, including primary containers and closures. It is essential that sterile equipment is used in the processing of all elements of the sterile product. Conducting only cleaning and disinfection (e.g., (b)(4)) of such equipment is inappropriate, as it is not equivalent to use of (b)(4) (or equivalent sterilization method, such as (b)(4)) sterilization of equipment. Validation of sterilization cycles is based on sterility assurance evaluations that are comprised of rigorous physical and biological measurements. In addition, it is essential that only sterile instruments are used for aseptic processing line manipulations and in handling of sterilized materials. Between uses, sterile instruments are held under ISO 5 conditions and continuously maintained in a manner that prevents contamination. It is not acceptable to touch the sterile (b)(4) during aseptic production in the manner performed by your personnel. Annex 1 requires us to ensure that all product contact components and equipment should be sterilized prior to use. This includes raw materials and intermediates and/or bulk solutions, (clause 8.11). The sterilization process should be validated, both physically, with temperature sensors and where appropriate biologically with a suitable biological indicator challenge. The whole of the load should be subject to the required sterilant exposure , (clause 8.36). To ensure this and to deliver a reliable and repeatable sterilisation process the whole of the load should equilibrate to temperature before the sterilization exposure phase commences, (clause 8.52). If it is not possible to assemble or connect load items aseptically within the Grade A core (Grade B background) then sterilization in place validation process needs to be considered. Preferably intrinsic sterile connection devices should be designed to mitigate risk of contamination, (clause 8.14). For materials, items, or components necessary for aseptic processing, that are not direct/indirect product contact then validated disinfection and transfer protocols should be in place, they should be protected from recontamination and the pathways for potential contamination should be clearly risk assessed and included within the EM programme, (clause 8.49) Where items are sterilized in sealed packaging, the packaging must be compatible with the load items and the sterilization process. The sealing process should be validated to ensure integrity post sterilisation. Validation should consider integrity of the sterile barrier system and the maximum hold time prior to sterilisation established and the shelf life assigned to each load item or component should be established, (clause 8.48). Maximum holding times for equipment and component cleaning, drying and sterilization should be documented and validated. The maximum holding time for sterilized equipment and components can be stored prior to use and during filling should be established and validated, (clause 8.18) & (clause 8.46) The qualification of the loading configuration and of the wrapping, orientation and location of individual load items should be carefully considered and clearly documented, such that their unwrapping to enable assembly with other direct and/or indirect product contact parts can be achieved aseptically, (clause 8.12). For moist heat porous hard goods loads, items to be sterilized must be clean, dry and packaged into their protective barrier systems, that comply to relevant parts of ISO 11607. Orientation to allow effective air removal and rapid steam penetration should be ensured, (clause 8.61). Equally, condensate drainage and confirmation the load items are ‘sensibly dry’ upon cycle completion should be verified both in qualification of the load and in routine operation, (clause 8.56).

0 Warning Letter - Failure to define Load Configurations
I like to keep an eye on FDA Warning letters, especially for anything related to sterilization process or equipment issues. This one has recently popped up, dated 5th June 2024. 1. Failure to establish and follow appropriate written procedures designed to prevent microbiological contamination of drug products purporting to be sterile, including procedures for validation of all aseptic and sterilization processes [21 CFR 211.113(b)]. For example: a. The aseptic processes used to manufacture your products have not been validated (e.g., by performing media fill simulations). These products purport to be sterile and are expected to be sterile. b. The (b)(4) sterilization process used for equipment that comes into direct contact with your products has not been adequately validated. For example, you have not defined the optimal parameters and conditions, including the load configuration for sterilizing equipment such as the small instruments (e.g., forceps), glass beakers, and sometimes PPE, used in aseptic processing of your products. This for me is really an own goal for this company, (based in the US). GMP expectations in this matter are quite clear: - In PDA Technical Report No.01, (section 4.4.1.3) it requires porous load patterns to be established after OQ prior to beginning PQ, ensuring items are loaded within the useable chamber space and orientated to aid air removal, steam penetration and condensate drainage, etc. Loading instructions should be documented. In the EU, Annex 1, validated loading patterns should be established for all sterilisation processes and load patterns should be subject to periodic revalidation. Maximum and minimum loads should also be considered as part of the overall load validation strategy, (clause 8.38). Routine operating parameters should be established and adhered to for all sterilisation processes, e.g. physical parameters and loading patterns, (clause 8.40) There should be mechanisms in place to detect a sterilisation cycle that does not conform to the validated parameters. Any failed sterilisation or sterilisation that deviated from the validated process (e.g. have longer or shorter phases such as heating cycles) should be investigated, (clause 8.41). Need help in defining and qualifying your sterilization load configurations? Worried you may not comply. Reach out to me here and avoid an audit observation.

0 NSAI - Working Group 3 on ISO 19253
It was great to attend the NSAI plenary event last week and work on ISO 19253, the new standard for moist heat sterilization of contained products. The plenary event is taking place from June 24th to June 28th at the Atlantic Technological University Galway City Campus. ISO/TC 198 is responsible for specifying requirements for #cleaning, #disinfecting, #sterilizing and aseptic processing of #health care products together with associated equipment and ancillary products used in ensuring effective application of these processes. ISO TC 198 is also responsible for harmonizing terminology related to its scope of work. This event will see the gathering of #global #experts in the field in the pursuit of the further development of standards in this area.Read more about the event here - https://lnkd.in/ehwVWmQB
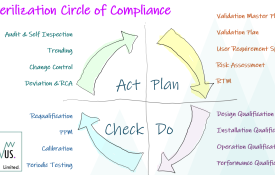
0 Sterilization Process & Equipment Validation: Circle of Compliance
Many are familiar with the Validation V-Model and the Deming Wheel. Like the Reusable Instrument Cycle in HTM01-01 [1] I like to visualize the Validation Lifecyle as a circular journey, moving through each of the 4 Stage Gates of Plan-Do-Check-Act. Within each stage there are discreet activities that we must complete and over the coming weeks I will go through each stage and discuss the essential elements that one must consider in the Commissioning, Validation, Routine Monitoring & Control of your sterilization process or equipment.For me, the validation exercise is not a journey along a one way street, culminating in annual Requalification testing, but a circle of compliance measures, checks and balances put in place to maintain our compliance position and sustain a repeatable and reliable service. [1] HTM01-01: Part A - Management & Provision, Clause 1.5.
0 New Publication: BS EN ISO 11607-1:2020+A1:2023
The importance of BS EN ISO 11607-1:2020+A1:2023.BS EN ISO 11607-1:2020+A1:2023 can help users to develop their expertise in terminal sterilization, while increasing confidence in their medical devices.This standard helps improve efficiency and strengthen their risk management, alongside safeguarding product quality and patient well-being. Navigating the complex regulatory landscape is a key aspect of sterilization validation. Kevin's in-depth knowledge of global regulations, including FDA, ISO, and industry-specific guidelines can assist you in developing effective moist heat sterilization validation strategies, preparing regulatory submissions, and ensuring compliance with the applicable standards. With his guidance, you can navigate the regulatory processes smoothly and efficiently, saving time and resources whilst meeting the necessary requirements.View this standard on BSI Knowledge today, click below for more.#BSIStandards #BSENISO11607 #MedicalDevices
0 New publication: BS EN ISO 17665:2024
Moist heat sterilization just got easier. BS EN ISO 17665:2024 is the newly published and radically revised international standard on steam sterilization processes for medical devices. It’s now fully up to date and utterly comprehensive.To comprehend the major changes, read our free of charge Executive Briefing. It breaks down BS EN ISO 17665:2024 into bite-sized, actionable insights that explains what the standard covers, and the changes that have been made, challenges and benefits of using it. Navigating the complex regulatory landscape is a key aspect of sterilization validation. Kevin's in-depth knowledge of global regulations, including FDA, ISO, and industry-specific guidelines can assist you in developing effective moist heat sterilization validation strategies, preparing regulatory submissions, and ensuring compliance with the applicable standards. With his guidance, you can navigate the regulatory processes smoothly and efficiently, saving time and resources whilst meeting the necessary requirements. Download now: https://bit.ly/3ycqOqxOr shop today: https://bit.ly/4bnU0cy#BSIStandards hashtag#ISO17665 #MedicalDeviceSterilization #MoistHeatSterilization #MedicalDevices
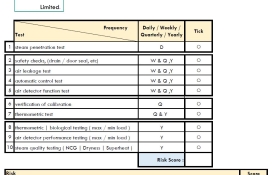
0 Annex 1 - are you in Control?
Annex 1 lists the essential expectations. However, to mitigate risk into the green zone, a more robust and integrated sterilization process control strategy is recommended, based upon sound and long established best practices. Inspecting or auditing a porous load autoclave? How robust is the autoclave control strategy? Complete my check list and determine the risk score? If the score is in the red or low orange the sterilization process may be at risk. Sterilization is a core pillar in the contamination control strategy; A quick assessment using the 10-point checklist below is recommended, which helps determine how robust the routine monitoring and control of the autoclave is. Periodic steam penetration tests, air leakage testing, annual requalification of load patterns and steam supply testing is not enough. More is required to provide assurance that the process continues to perform as validated and within defined permitted limits. Data is key in being able to trend autoclave performance. The recommended periodic tests have been there for many years in the UK and in the standards, and we should not be selective in what we do or do not do. As I have mentioned in recent technical notes on Annex 1 changes, the required tests are mutually dependent to provide a clear picture to the user of the ongoing performance of the autoclave. If you are selective in the testing performed, the full and complete picture is not apparent and therefore the early detection of a risk/problem as a consequence is reduced. Zimmermann recently said at the ISPE Aseptic Conference: - “Know what you do and why, and what the consequences might be if you would not do it”. ISPE Aseptic Conference. Choosing to not follow long established best practice has consequences when it comes to understanding process failure, determination of root cause and effective CAPA. Understanding the gaps in your control strategy and risks for contamination to enter your process is key. Identify what you are doing; you may have a sterilisation process that works but you must understand how it works and where the boundaries are and of the critical control points that maintain a safe and effective sterilization service. You need the collective data generated from the full and complete set of periodic testing in order to demonstrate performance and control, not just annually but daily and weekly. Sterilization process control, key points to consider: - 1.Integrity of data. The nature and frequency of periodic tests, sufficient to give an overall view of the sterilization process performance. Not performing tests in isolation by differing stakeholders, but as part of an integrated testing strategy, performed and reported by a competent, skilled and knowledgeable test person. 2. Data Recording. Simple standardized forms, designed to capture the most relevant sterilization process data are recommended, allowing the test person to collate and present the data in a transparent and meaningful format that demonstrates continued performance and compliance. 3. Data evaluation. Who is evaluating the data? Are they trained and qualified to perform the tests, to review the data and is the person checking the data an appropriately skilled and qualified person? Quality oversight and approval by a sterilisation SME who has demonstrable knowledge, experience and training is highly recommended. Collectively the individuals in the chain of responsibility should be able to identify and pre-empt potential issues before they manifest and be able to confidently dismiss risk to product quality or patient safety. This can only be assured where the correct testing and their frequencies are being performed. 4. Trend analysis. Data from a single test is often not significant, (a satisfactory annual Requalification test is not confirmation that the autoclave has performed within acceptable limits for the previous 12months). Furthermore, sterilization processes will have a degree of variability, (whilst operating within defined and validated permitted tolerances). Therefore, graphic presentation of results collected over time should be considered, to distinguish cycle variation from trends or in indicating a significant change in process has occurred, even though the data may fall within specified permitted tolerances. Thank you for reading and please check out my recent technical notes on the subject of Annex 1 changes here: - Technical Note 01 - Annex 1 - Sterilization - 8.36, 8.52, 8.57, 8.58 & 8.59 Technical Note 02 - Annex 1 - Load Patterns - 8.38, 8.39, 8.40 & 8.61 Technical Note 03 - Annex 1 - Automatic Control Test - 8.35 & 8.41 Technical Note 04 - Annex 1 - Air Detecting Systems - 8.61 If you are about to conduct a self inspection or wish to discuss further your test strategy and how to report your data, then contact me directly at the following email address: - KJP@pavus.co.uk Thanks,